
透析患者の貧血管理において、
「ESAを増やしてもヘモグロビンが上がらない」
「鉄は足りているはずなのに反応が悪い」
そんな説明しきれない“もどかしさ”を感じたことはないでしょうか。
一方で、CKD-MBDの管理では、リン、副甲状腺ホルモン(PTH)、ビタミンD、FGF-23といった指標を日常的に追いながらも、それらが貧血とどう結びついているのかを体系的に理解する機会は多くありません。
近年、FGF-23は単なる「リン調節ホルモン」ではなく、
貧血・炎症・鉄代謝・ESA低反応性にまで深く関与する因子であることが明らかになってきました。
さらに、腎臓だけでなく骨・骨髄を巻き込んだ
「腎臓―骨髄―骨軸(kidney–bone marrow–bone axis)」
という新しい概念が、CKDにおける貧血とMBDを一つの連続した病態として説明し始めています。
本記事では、最新のレビュー論文をもとに、
FGF-23を軸としてCKD貧血とCKD-MBDがどのようにつながっているのかを整理し、
ESA抵抗性、鉄管理、HIF-PHI、PTH・Klothoといった要素を
「臨床でどう読み替えるか」という視点で解説します。
- CKD貧血とCKD-MBDは「別の病態」ではない
- FGF-23とは何者か?
- FGF-23はなぜ貧血を悪化させるのか
- EPOとFGF-23の相互作用
- CKDにおける「腎臓―骨髄―骨軸」
- 鉄・HIF-PHI・PTH治療をどう再解釈するか
- 鉄補充療法
- HIF-PHI(HIFプロリル水酸化酵素阻害薬)
- 続発性副甲状腺機能亢進症(PTH管理)
- この章のまとめ
- 明日からの透析現場で何を見るか(超実践編)
- 1)ESAが効かない時の「3ステップ問診(現場版)」
- 2)FGF-23を測っていない施設でも“近いもの”は見られる
- 3)「鉄の入れ方」を戦略にする
- 4)ESAかHIF-PHIか:判断軸を「Hb」から「環境」に寄せる
- 5)超・実用「ESA低反応性」ミニアルゴリズム
- 6)このレビューを現場で使う「最短の結論」
- まとめ
- あとがき
- 翻訳全文
CKD貧血とCKD-MBDは「別の病態」ではない
慢性腎臓病(CKD)の合併症として、貧血と””骨・ミネラル代謝異常(CKD-MBD)””は、これまで「別々の問題」として扱われてきました。
貧血はEPO(エリスロポエチン)と鉄の問題、CKD-MBDはリン・カルシウム・PTH・ビタミンDの問題——。
臨床現場でも、多くの場合はそのように整理されているはずです。
しかし実際の透析医療では、
- ESAを増量してもHbが上がらない
- 鉄は補充しているのに反応が悪い
- PTHやリン管理が乱れている患者ほど貧血も難治
といった 「説明しきれない重なり」 を日常的に経験します。
この違和感に対して、近年の研究は
「貧血とCKD-MBDは同じ病態ネットワークの中にある」
という見方を示し始めています。
その中心に位置する分子が FGF-23 です。
FGF-23とは何者か?
― リンのホルモンを超えた存在 ―
FGF-23(Fibroblast Growth Factor-23)は、もともと
血清リン濃度を一定範囲に保つためのホルモン
として知られてきました。
骨細胞・骨芽細胞から分泌され、
- 腎臓でのリン再吸収を抑制
- 活性型ビタミンD(カルシトリオール)産生を抑制
することで、リン負荷から身体を守ります。
重要なのは、FGF-23は
血清リンやPTHが異常を示すよりもはるかに早期、
CKDの初期段階から上昇し始める点です。
つまりFGF-23は、
CKD-MBDの「結果」ではなく「最初に動く因子」
だと言えます。
さらに近年、FGF-23はリン代謝だけでなく、
- 炎症
- 鉄代謝
- 貧血
- 心血管イベント
- 予後
にまで深く関与していることが分かってきました。
FGF-23はなぜ貧血を悪化させるのか
FGF-23が貧血と関係する理由は、一つではありません。
むしろ複数の経路を通じて、間接的に造血を妨げるのが特徴です。
炎症とヘプシジンを介した作用
FGF-23は、全身性炎症と強く関連します。
炎症が持続すると、肝臓でヘプシジンが産生され、
- 鉄が骨髄マクロファージ内に隔離される
- 腸管からの鉄吸収が抑制される
結果として、
「鉄はあるのに使えない」=機能的鉄欠乏
が生じます。
この状態では、ESAを投与しても赤血球造血は効率よく進みません。
FGF-23はESA低反応性の背景因子
実際、臨床研究では
FGF-23高値とESA低反応性の関連が示されています。
興味深いのは、
FGF-23は「高すぎても」「低すぎても」ESAが効きにくい
という U字型の関係 を示す点です。
- 高FGF-23:炎症・鉄利用障害・造血抑制
- 低FGF-23:低栄養・骨代謝低下・骨髄の省エネ状態
どちらも、造血にとっては不利に働きます。
EPOとFGF-23の相互作用
― 骨髄が主役になる瞬間 ―
このレビューで最も重要なポイントの一つが、
EPOとFGF-23の双方向性です。
EPOはFGF-23を増やす
ESA(rhEPO)を投与すると、
- 骨髄でFGF-23の転写が誘導され
- 主に「切断型FGF-23(cFGF-23)」が増加
することが、動物実験・ヒト研究の両方で示されています。
特筆すべきは、
FGF-23が骨だけでなく「骨髄」で作られる
という事実です。
FGF-23はEPOを抑える
一方で、FGF-23を外から投与すると、
- 腎臓でのEPO遺伝子発現が低下
することも確認されています。
つまり、
EPO → FGF-23↑ → EPO↓
という 負のフィードバックループ が存在します。
これが、
「ESAを増やしても、どこかで頭打ちになる」
理由の一つだと考えられます。
CKDにおける「腎臓―骨髄―骨軸」
これらを統合すると、CKDでは
- 腎臓:EPO産生低下
- 骨:FGF-23上昇
- 骨髄:造血抑制・FGF-23産生
が相互に影響し合う
「腎臓―骨髄―骨軸」 が形成されます。
貧血も、CKD-MBDも、ESA低反応性も、
この軸の中で同時に進行する現象なのです。
鉄・HIF-PHI・PTH治療をどう再解釈するか
―「何を入れるか」より「何が動くか」を考える ―
CKD貧血の治療では、
鉄補充・ESA・HIF-PHI・PTH管理
といった複数の介入を同時並行で行うことが珍しくありません。
しかし本レビューが示しているのは、
それぞれの治療がFGF-23を介して“別の場所にも影響を及ぼしている”
という事実です。
ここでは、従来の治療を
FGF-23という視点で再解釈してみます。
鉄補充療法
―「Hbを上げる治療」から「FGF-23を整える治療」へ ―
鉄補充は、貧血治療の基本です。
ただしCKDでは、多くの患者が
- 血清鉄は低くない
- フェリチンも極端に低くない
にもかかわらず、造血が進まない状態に陥ります。
これは「鉄が足りない」のではなく、
**鉄が使えない(機能的鉄欠乏)**状態です。
FGF-23と鉄の意外な関係
鉄欠乏(特に機能的鉄欠乏)は、
- HIF活性化
- FGF-23転写の亢進
- FGF-23切断の増加
を引き起こします。
つまり鉄欠乏は、
貧血を悪化させるだけでなく、FGF-23をさらに上げる因子
でもあります。
経口鉄と静注鉄の「違い」
興味深いのは、
- 経口鉄:cFGF-23・iFGF-23ともに低下
- 静注鉄:cFGF-23は低下するが、iFGF-23が上昇する場合あり
という報告がある点です。
これは
「鉄を入れれば同じ」
ではないことを示しています。
FGF-23の観点から見ると、
鉄は“入れ方”まで含めて治療
と考える必要がありそうです。
HIF-PHI(HIFプロリル水酸化酵素阻害薬)
― ESAの代替ではなく「病態修飾薬」 ―
HIF-PHIは、
- 内因性EPO産生を促進
- ヘプシジンを低下
- 鉄利用効率を改善
する新しい貧血治療薬です。
そのため、
「ESAより生理的」
「鉄が効きやすくなる」
というイメージで語られがちです。
しかしFGF-23の視点では…
一部の研究では、HIF-PHI(特にロキサデュスタット)使用により、
- FGF-23(主にcFGF-23)が増加
することが示されています。
これは、
- HIF活性化
- EPO転写誘導
- 骨髄でのFGF-23産生
という経路が同時に走るためと考えられます。
一方で、臨床研究では
- EPO濃度は必ずしも上昇しない
- ヘプシジンは低下
- 鉄利用は改善
という結果も報告されており、
FGF-23が上がる=必ず悪い
とは言い切れません。
重要なのは、
HIF-PHIは「Hbを上げる薬」ではなく、
造血環境全体を動かす薬である
という理解です。
続発性副甲状腺機能亢進症(PTH管理)
― 骨の問題は、実は骨髄の問題でもある ―
PTHは、CKD-MBD管理の中心的指標です。
しかしPTHが高い状態が続くと、
- 破骨細胞活性の持続
- 骨リモデリング亢進
- 骨髄線維化
が進行します。
骨髄線維化=造血の場が壊れる
骨髄線維化が進むと、
- 赤血球系前駆細胞が増えにくくなる
- ESA反応性が低下
します。
つまりPTH管理は、
骨折予防だけでなく「貧血治療の前提条件」
でもあります。
シナカルセトが貧血を改善する理由
臨床研究では、
- シナカルセト投与
- Hb上昇
- ESA必要量減少
- FGF-23低下
が同時に起こることが示されています。
これは、
- PTH低下
- 骨リモデリング正常化
- 骨髄環境改善
- FGF-23抑制
という連鎖の結果と考えると、非常に納得がいきます。
この章のまとめ
― 治療を「点」ではなく「ネットワーク」で考える ―
鉄、HIF-PHI、PTH治療は、
それぞれ独立した対策ではありません。
すべてが
- FGF-23
- 骨
- 骨髄
- 炎症
- 鉄利用
を介して、互いに影響し合っています。
これからのCKD貧血管理では、
「Hbをどれだけ上げるか」ではなく
「造血が起こりやすい環境をどう整えるか」
という視点が、より重要になるはずです。
明日からの透析現場で何を見るか(超実践編)
― ESAが効かない時に“次に見るべきもの”チェックリスト ―
0)まず大前提:Hbだけ見てると迷子になります
ESAの効きが悪いとき、つい「増量」か「鉄追加」に寄りがちです。
でも本レビューの視点では、””造血不良の背景はネットワーク(炎症・鉄利用・骨髄環境・PTH・FGF-23)””です。
なので手順はこう:
① 造血の材料(鉄)
② 造血のブレーキ(炎症・ヘプシジン)
③ 造血の“畑”(骨髄=PTH/栄養)
④ 薬の方向(ESA/HIF-PHI/鉄の入れ方)
の順で見ていきます。
1)ESAが効かない時の「3ステップ問診(現場版)」
ステップ1:鉄は“足りてる”ではなく“使えてる”?
- TSAT(移送鉄):低いならまず鉄供給不足寄り
- フェリチン:高いのにTSAT低いなら「機能的鉄欠乏」疑い
- CRPが高いと、鉄は骨髄に“封印”されやすい
現場の読み替え:
フェリチンが高い=鉄が十分、ではなく
「炎症で鉄が倉庫に閉じ込められている」可能性あり。
ステップ2:炎症の火種があるか?(CRPだけで終わらせない)
CRPが高い=ESA低反応性の定番ですが、さらに
- 透析アクセス感染、歯科、皮膚、慢性創傷
- 透析膜・透析液・透析条件による慢性炎症
- 栄養低下(MIA:低栄養‐炎症複合)
ここを拾えると、ESA増量より先にやることが見えてきます。
ステップ3:PTHは「骨の指標」で終わってないか?
PTH高値が長期化すると、骨リモデリングの暴走 → 骨髄線維化 → 造血低下、という文脈が出てきます。
- PTH高値+ALP高め+Hb反応悪い
→ “骨髄の畑が荒れている”可能性
介入の方向:
- 副甲状腺機能亢進の是正(例:カルシミメティクス)
- リン管理・活性型VitD調整
が「貧血改善の一手」になり得ます。
2)FGF-23を測っていない施設でも“近いもの”は見られる
FGF-23は多くの施設で日常測定ではありません。
でも代わりに、FGF-23が上がりやすい状況は拾えます。
「FGF-23が上がりやすい」臨床の匂い
- リン高値(または“リンが正常でもPTH/VitDが崩れてる”)
- 炎症が続いている
- 鉄欠乏(絶対/機能的)
- ESA高用量になっている
- HIF-PHI導入・切替直後(主にcFGF-23が増えやすい可能性)
ここを押さえるだけで、臨床の打ち手が整理されます。
3)「鉄の入れ方」を戦略にする
このレビューの示唆は強烈で、
- 経口鉄:cFGF-23/iFGF-23が下がりやすい報告
- 静注鉄:cFGF-23は下がっても、iFGF-23が上がる報告あり
という“差”がありました。
明日からできる現場の問い:
- この患者は「追加」より「使える環境作り(炎症/PTH)」が先では?
- IV鉄を入れ続けているが、背景に慢性炎症が残っていないか?
- リン吸着薬が鉄含有(クエン酸第二鉄など)なら、貧血とMBDを同時に動かしていないか?
4)ESAかHIF-PHIか:判断軸を「Hb」から「環境」に寄せる
HIF-PHIは
- ヘプシジン低下
- 鉄利用改善
が期待でき、ESAとは別ベクトルの強みがあります。
ただし、FGF-23(主に切断型)が動く可能性も示唆されています。
現場の再解釈:
- Hbが上がる/上がらないだけでなく
- TSAT/フェリチン/CRP/PTH/リンがどう動いたか
で評価すると、治療が“迷走”しにくいです。
5)超・実用「ESA低反応性」ミニアルゴリズム
- Hb低下 → まず出血・溶血・採血頻回・透析条件ミスを除外
- TSAT×フェリチン×CRPで鉄利用を評価
- TSAT低い:供給不足寄り
- フェリチン高+TSAT低+CRP高:機能的鉄欠乏寄り
- PTH/ALP/リンで骨髄環境(骨代謝)を評価
- 介入の優先順位を決める
- 炎症源の是正 → 鉄利用改善
- PTH是正 → 骨髄環境改善
- 鉄の入れ方最適化
- ESA/HIF-PHIの選択・調整
6)このレビューを現場で使う「最短の結論」
ESAが効かない=薬が弱いのではなく、造血の環境が悪い。
その環境は、””炎症(ヘプシジン)・鉄利用・PTH/骨髄・リン/VitD(FGF-23)””で説明できる。
まとめ
― CKD貧血を「FGF-23を軸に再構築する」 ―
透析患者の貧血は、
EPOが足りないから、
鉄が足りないから、
それだけでは説明できません。
本記事で見てきたように、CKDでは
- FGF-23
- 炎症(ヘプシジン)
- 鉄利用障害
- 骨代謝(PTH)
- 骨髄環境
- EPO/ESA/HIF-PHI
が互いに影響し合う
“”「腎臓―骨髄―骨軸」””の中で、貧血が進行します。
✔ 本記事の要点(Take Home Message)
- FGF-23はリンのホルモンにとどまらず、貧血・炎症・ESA低反応性をつなぐハブ
- ESAが効かない原因は「用量不足」ではなく「造血環境の悪化」であることが多い
- 鉄は「あるか」ではなく「使えているか」を見る(機能的鉄欠乏)
- PTH管理は骨折予防だけでなく、骨髄環境=造血の土台を整える治療
- EPOはFGF-23を増やし、FGF-23はEPOを抑える負のフィードバックを持つ
- HIF-PHIはHbだけで評価せず、鉄利用・炎症・PTH・リンの動きをセットで見る
✔ 明日からの臨床での問い
ESAを増量する前に、こう問い直してみてください。
- 鉄は足りているのか?それとも使えていないのか?
- 炎症や低栄養という見えにくいブレーキはかかっていないか?
- PTH高値によって、骨髄の“畑”が荒れていないか?
- 貧血とMBDを、別々に治療していないか?
✔ 最後に
CKD貧血の治療は、
“”「Hbを上げる作業」ではなく、「造血が起こりやすい環境を整えるプロセス」””です。
FGF-23という視点を加えることで、
これまで点で見ていた
貧血・リン・PTH・鉄・ESAが、一本の線でつながります。
透析室で感じる
「なぜこの患者はうまくいかないのか?」
その答えは、””検査値の裏側にある“ネットワーク”””にあります。
あとがき
さーてさて。今回はレビュー論文をざざっと記事にしてみました。
論文自体がとてもビックバリューなため、大分長文となりました。
しかし、今回の主役であるFGF-23についての知見は大変深まったのではないでしょうか。
Anemia-MBD-boneの連関が、こうも複雑かつ多面的に絡んでいると、理解はとても難しいものとなるのではないでしょうか。
そんな皆様の疑問に答えれる記事になったのではないでしょうか。
是非とも、今回の考察を現場に持ち帰り、データを俯瞰してみてください!!
翻訳全文
Anemia and Mineral Bone Disorder in Kidney Disease Patients:The Role of FGF-23 and Other Related Factors
腎疾患患者における貧血とCKD-MBD:FGF-23および関連因子の役割
抄録(Abstract)
貧血および骨・ミネラル代謝異常(mineral and bone disorder:MBD)は、慢性腎臓病(CKD)における重要な合併症です。エリスロポエチン(Epo)経路は、CKDにおけるこれら両者の病態において中心的な役割を果たしています。
一方、CKD-MBDにおいて重要な役割を担うもう一つの分子が線維芽細胞増殖因子23(fibroblast growth factor-23:FGF-23)です。FGF-23の主な役割は、共受容体であるKlothoを介して血清リン濃度を正常範囲内に維持することですが、その活性は貧血や炎症とも関連している可能性があります。
本総説では、EpoおよびFGF-23の調節機構と、それらの分子レベルでの作用機序について概説します。さらに、EpoとFGF-23の複雑な相互作用について議論するとともに、Klotho、ビタミンD、鉄欠乏など、貧血に関連する他の因子やプロセスとの関連性についても取り上げます。
これらの要素は総合的に、「腎臓―骨髄―骨軸(kidney–bone marrow–bone axis)」の一部を形成し、CKD-MBDの病態形成を促進している可能性があります。
Keywords: chronic kidney disease; mineral bone disorder; erythropoietin; ESKD; Klotho; anemia
1. はじめに(Introduction)
エリスロポエチン(erythropoietin:Epo)は、成熟赤血球の産生に至る複雑な増殖および分化過程において、中心的な役割を担うタンパク質と考えられています。しかし近年の研究により、Epoには多彩なパラクリン作用および内分泌作用が存在することも報告されています。実際に、神経細胞、心筋細胞、血管内皮細胞や血管平滑筋細胞など、さまざまな細胞種がEpoを産生し、かつ細胞膜表面に特異的なEpo受容体を発現していることが示されています。
慢性腎臓病(chronic kidney disease:CKD)において、Epo経路は貧血の発症に関与するだけでなく、生化学的異常、骨障害、血管石灰化を特徴とする全身性疾患であるCKD関連骨・ミネラル代謝異常(CKD-mineral and bone disorder:CKD-MBD)の制御にも関与していると考えられています。CKD-MBDにおける重要な分子の一つが、リン代謝の主要な調節因子である線維芽細胞増殖因子23(fibroblast growth factor-23:FGF-23)です。
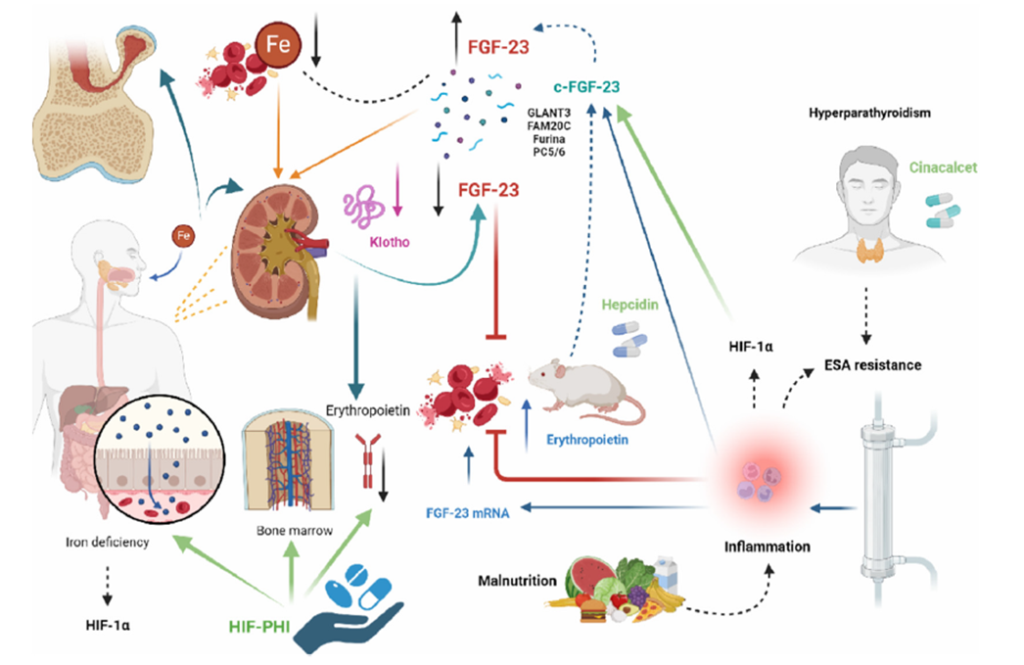
本ナラティブレビューの目的は、貧血および貧血関連因子、FGF-23、ならびにCKD-MBDの病態形成に関与するその他の分子との間に存在する、新たに明らかになりつつある複雑な相互関係を概説し、「腎臓―骨髄―骨軸(kidney–bone marrow–bone axis)」(図1)として定義することにあります。本論文では、FGF-23およびビタミンDが鉄代謝やCKDに続発する貧血の発症において果たす重要な役割についても明らかにします。
さらに、炎症や鉄欠乏の状態と同様に、EpoとFGF-23の間にはフィードバック機構が存在することが示されています。FGF-23は、Epoによる刺激を受けて骨髄でも産生されることが知られています。また、FGF-23の共受容体であるKlothoも、この「軸」における重要な構成要素であり、その血中濃度はCKDの早期段階から低下し、その後Epo刺激により増加することが報告されています。
2. 腎臓―骨髄―骨軸に関与する因子
2.1. EPO:合成・調節機構・シグナル経路
ヒトのエリスロポエチン(Epo)は、循環血中では分子量30.4 kDaの糖タンパク質ホルモンとして存在し、165個のアミノ酸から構成され、4つの糖鎖が結合しています。胎児期においてはEpoは主に肝臓で産生されますが、成人では腎臓が主要な産生臓器となり、腎皮質中部に存在する尿細管周囲間質線維芽細胞によって産生されます。この領域は酸素消費量が高く、かつ酸素供給が少ないという特徴を有しており、低酸素状態がEpo産生の主要な刺激となります。尿細管周囲毛細血管の内皮細胞は酸素分圧のセンサーとして機能し、酸素濃度に応じてEpoを産生する線維芽細胞の数が調節されます[1,2]。
Epo産生を制御する主要な調節変数は組織酸素分圧(pO₂)であり、これはヘモグロビン(Hb)濃度、動脈血pO₂、Hbの酸素親和性と密接に関連しています。腎皮質領域のpO₂は血流量の変化による影響を受けにくいため、腎臓は酸素依存性のEpo産生に適した臓器といえます[1,3]。
Epo遺伝子(EPO)は第7染色体上に存在し、その転写調節は複数の転写因子によって制御されています。低酸素によるEPO転写誘導を担う主な転写活性化因子は、低酸素誘導因子(hypoxia-inducible factors:HIFs)と呼ばれるヘテロ二量体転写因子です[4,5]。HIFは酸素感受性のαサブユニット(HIF-1α、HIF-2α、HIF-3αの3種類)と、恒常的に発現しているβサブユニット(aryl hydrocarbon receptor nuclear translocator:ARNT)から構成されます[6–9]。Epoの転写は主としてHIF-2によって制御されており、HIF-1βとHIF-2αからなる複合体が関与します[10]。HIF-1およびHIF-3は、低酸素応答配列(hypoxia response elements:HREs)と呼ばれる特異的DNA配列に結合することで遺伝子発現を調節します[11]。
HIF活性の調節は主にプロリル水酸化酵素(prolyl-hydroxylases:PHD)によって行われており、これらは血中の鉄、酸素、α-ケトグルタル酸、アスコルビン酸(ビタミンC)、および活性酸素種(ROS)によって活性化されます[12]。
さらに、HIF-1αを水酸化し、補因子との結合を阻害することで転写を抑制する因子として、アスパラギニル水酸化酵素(factor-inhibiting HIF:FIH)も存在します[13]。常酸素状態(酸素供給が需要を上回る状態)では、HIFは酸素依存的に継続的に分解されるため、標的遺伝子の転写を誘導できません[12,14]。一方、低酸素条件下では、酸素分圧の低下によりPHDおよびFIHの活性が低下し、HIF-1αが分解されずにHIF-1βと二量体を形成します。この複合体は核内へ移行し、CBP(CREB binding protein)/P300の存在下で低酸素応答遺伝子の転写を活性化します[15]。この一連のカスケードによりEPOが活性化され、酸素分圧が再び上昇するとHIF-1αは再度分解されます。
HIFのもう一つの重要な役割は、鉄代謝および酸素輸送に関与する遺伝子の転写調節です。具体的には、HIF-1はトランスフェリン、セルロプラスミン(フェロキシダーゼ)、トランスフェリン受容体1をコードする遺伝子の転写を促進します。HIF-2はHIF-1活性を調節するとともに、小腸における二価陽イオン輸送体やフェロポルチンの合成を増加させ、鉄の吸収および造血部位への輸送を促進します[16]。
Epo受容体(EpoR)はキナーゼ活性を持たないサイトカイン受容体スーパーファミリーに属する膜貫通型受容体であり、赤芽球系前駆細胞に発現しています。また、前述のとおり、造血系以外の組織にも存在します。ヒトの内皮細胞、腎細胞、心筋細胞、神経細胞におけるEpoR mRNA発現量は、Epoに高感受性な細胞と比べて10~100分の1程度とされています[17,18]。
骨髄において最も多くEpoRを発現している細胞は、赤芽球コロニー形成単位(CFU-E)および前赤芽球です。一方、成熟赤血球へと分化が進むにつれてEpoRの発現は徐々に低下し、網状赤血球および成熟赤血球にはEpoRは存在しません[1]。
EpoがEpoRに結合すると、受容体のホモ二量体化が起こり[19]、これに続いてJanusキナーゼ(JAK)ファミリーに属するチロシンキナーゼ、特にJAK2が活性化されます[20]。リン酸化されたEpoRは細胞内シグナルカスケードを活性化し、抗アポトーシス作用、分化促進作用、増殖促進作用を発揮します。実際に、腎細胞障害や細胞死を誘導するさまざまな有害刺激を用いたin vitroおよびin vivoモデルにおいて、Epoの保護作用が示されています[21–26]。
Epo mRNAは骨髄、脾臓、肝臓、胎盤、脳、生殖器、肺など、他の臓器でも検出されています[1,3,27–29]。これらの組織では、Epoは赤血球産生以外の機能に関与しています。
脳は低酸素に応答してEpoを産生できることが示されており、とくに脳室周囲領域やアストロサイトでその産生が認められます[30]。中枢神経系におけるEPO発現は、正常な脳発達に重要であると考えられており[31]、神経保護作用[32–35]や脳血流調節[36]に関与している可能性があります。脳および精巣におけるEPO発現の共通点は、血液脳関門および血液精巣関門によって全身循環から隔離されている点であり、内分泌作用というよりもパラクリン作用として機能していることを示唆します[28,37,38]。ただし、脳損傷など特定の条件下では、Epoが血液脳関門を通過する可能性も示されています[35]。
女性生殖器に関しては、ラットにおいて子宮[39]および卵管[40]でEPO発現が確認されており、ヒトでは月経周期に関連した変動を伴って子宮内膜にEPOが発現することが示されています[41]。Epo mRNAは胎盤の胎児側および母体側の両方で発現しており[27,42,43]、その機能は明確ではないものの、消化管に対する栄養的(トロフィック)作用を有すると考えられています[44]。
さらに、高感度mRNA検出法により、肺、脾臓、心臓、骨髄におけるEPO発現も報告されています[45]。骨髄では、循環Epoほど顕著ではないものの、自己分泌および/または傍分泌機構を介して赤血球造血に関与していると考えられています。これは、腎不全に起因する腎性貧血の存在によって裏付けられています[3]。心臓におけるEPO発現は低レベルですが、心臓形態形成に関与している可能性が示唆されています[46]。
2.2. CKD-MBDと貧血:異なる臨床的表現型
CKD関連骨・ミネラル代謝異常(CKD-MBD)は、慢性腎臓病(CKD)の臨床スペクトラムの中でも、予後に最も大きな影響を与える合併症の一つです。カルシウム、リン、副甲状腺ホルモン(parathyroid hormone:PTH)、FGF-23、ビタミンD代謝の異常に加え、骨代謝回転、骨の石灰化、体積および線形成長、骨強度、さらには骨外石灰化の異常がCKD-MBDの病態に関与しています[47–49]。
CKD-MBDに特徴的な変化は、腎機能の低下に伴って徐々に進行し、推算糸球体濾過量(estimated glomerular filtration rate:eGFR)が 40 mL/min/1.73 m²未満 になると臨床的に明らかとなります。ただし、これらの要素の一部は、血清カルシウム、リン、PTH、ビタミンD濃度に明らかな変化が認められる前の、CKD早期から出現することが知られています。具体的には、FGF-23分泌の増加、Klotho欠乏、骨形成速度の低下、血管石灰化などが早期から認められます[50–55]。
CKD-MBDの病態スペクトラムは、CKDの重症度だけでなく、腎障害の原因によっても異なります。腎性骨異栄養症(renal osteodystrophy)は、CKD-MBDにおける骨異常の一要素に過ぎず、骨生検によって確認されるCKDに伴う骨形態学的変化を指します。腎性骨異栄養症は主に4つのタイプに分類されます:
① 線維性骨炎(従来は線維性嚢胞性骨炎と呼ばれていた)、
② 骨軟化症、
③ 無形成骨疾患(adynamic bone disease)、
④ 混合型骨疾患、
です[47]。糖尿病性腎症患者において保存期治療を受けている場合、無形成骨疾患が明らかに優勢であることが示されています[56–58]。また、透析を受けている糖尿病患者では、非糖尿病患者と比較してPTHおよびアルカリホスファターゼの値が低いことも報告されています[59,60]。さらに、同じ糖尿病透析患者群では、非糖尿病患者よりも椎体骨折の有病率が高いことが示されています[56–61]。
常染色体優性多発性嚢胞腎(autosomal dominant polycystic kidney disease:ADPKD)患者においても、アルカリホスファターゼおよびPTHの低値、血中インタクトFGF-23(intact FGF-23:iFGF-23)の高値、骨形成速度の低下が認められています[62,63]。しかし、ADPKDは末期腎不全(ESKD)患者において骨折率の上昇とは関連していません[63]。
糸球体疾患患者、特にネフローゼ症候群を伴う症例では、骨障害の病因は極めて複雑かつ多様です。これは、ビタミンD恒常性への影響(ビタミンD結合タンパクの尿中喪失)、副腎皮質ステロイドやその他の免疫抑制薬の使用、炎症環境、免疫細胞および免疫因子の作用などが関与するためです。ネフローゼ症候群患者では、**ビタミンD濃度の低下に起因すると考えられる骨軟化症(高骨代謝を伴う場合も伴わない場合もある)**の高頻度な発生が報告されています[64]。この集団において、糸球体疾患が骨折リスク増加にどの程度寄与しているかについては、十分に検討されていません。
貧血の程度もまた、腎疾患の種類、進行段階、合併症の有無によって異なります。実際に、ADPKD患者では、他の原因によるCKD患者と比較して貧血の程度が軽いことが多く[65–68]、一部の患者ではヘモグロビン値が正常範囲に保たれています。Epo濃度は、他の原因によるESKD患者と比較して平均で約2倍高値を示します[69]。これは、嚢胞の進行性拡大によって血管が圧迫され、局所的な低酸素状態が生じ、HIFが活性化されるためと考えられています[70]。HIF-2αを介したEpo産生刺激は、ADPKD患者で他のCKD患者よりも貧血が軽度である理由を説明する要因と考えられています[69]。
2.3. FGF-23:産生と主な作用
FGF-23(線維芽細胞増殖因子23)は、食事性リン負荷の増加、カルシトリオール、副甲状腺ホルモン(PTH)、およびカルシウムによって刺激された骨組織内の**骨細胞(osteocyte)および骨芽細胞(osteoblast)**から産生されるホルモンです(図2)。FGF-23は腎臓、副甲状腺、心臓、骨、さらには他の臓器にも作用すると考えられています[71–80]。
FGF-23は、FGF-19およびFGF-21とともに、内分泌型FGFサブファミリーに属します[81]。FGFは4種類のFGFチロシンキナーゼ受容体(FGFR1~4)を介してシグナルを伝達し、RAS–MAPK経路およびPI3K–AKT経路を活性化します[82]。古典的なFGF-23シグナル伝達には、細胞外にグルクロニダーゼ活性を有する膜貫通型タンパク質であるα-Klothoが共受容体として必要であり、FGF受容体1c(FGFR1c)への結合を介して作用します[83,84]。一方で、FGF-23はKlotho非依存的にFGFR3およびFGFR4を活性化することでも作用し得ます[80,85]。
FGF-23の主な作用は、カルシトリオール産生の抑制を介して、腎臓および腸管におけるリン吸収を低下させ、血清リン濃度を正常範囲に維持することです[86]。FGF-23はリン恒常性の中心的調節因子であり、1,25-ジヒドロキシビタミンD(1,25(OH)₂D、カルシトリオール)に対する拮抗ホルモンとして機能します。
腎尿細管におけるFGF-23のリン排泄促進作用はKlotho依存性です。腎近位尿細管細胞では、FGF-23がFGFRおよび共受容体であるKlothoに結合し、管腔側膜に存在するナトリウム・リン共輸送体Na/Pi IIa(およびおそらくNa/Pi IIc)の発現を低下させます[87,88](図3)。
さらにFGF-23は、近位尿細管において25(OH)Dを活性型の1,25(OH)₂Dに変換する酵素である**1α水酸化酵素(CYP27B1)の発現を抑制し[89]、一方で25(OH)Dおよび1,25(OH)₂Dを不活性代謝物へ変換する24水酸化酵素(CYP24A1)**の発現を増加させます[90–92]。その結果、腎臓におけるカルシトリオール産生は低下し、最終的に血清リン濃度が低下します。
FGF-23の上昇は、血清カルシウム、リン、PTHに変化が現れる前から検出可能な、CKD-MBDの早期バイオマーカーです[54,55]。FGF-23濃度は末期腎不全(ESKD)発症の約5年前から上昇し始め、ESKD移行まで急速に増加し続けます[55]。FGF-23の進行性上昇は、CKD後期まで血清リン濃度を正常に保つのに寄与する一方で[54,76,93,94]、カルシトリオール産生抑制を介して続発性副甲状腺機能亢進症を促進します。
FGF-23の直接的な刺激因子としては、リポカリン2(lipocalin 2:LCN2;炎症マーカーであり鉄輸送体)[95]、低酸素状態、鉄欠乏、そしてEpoが挙げられます[96](図2)。
複数のCKD患者を対象とした臨床研究により、FGF-23の上昇は不良な予後と関連することが示されています[97–100]。さらに、Klotho欠乏は、FGF-23と死亡率あるいは心不全による入院との関連を交絡する因子ではないことが示されています[101]。
近年の科学的文献からは、カルシウム・リン代謝を制御する機構と赤血球造血を調節する機構との間に、ますます強い関連性が存在することが明らかになっています。後述するように、FGF-23は貧血および全身性炎症を促進する因子としても機能します。
2.3. FGF-23:産生と主な作用
FGF-23(線維芽細胞増殖因子23)は、食事性リン負荷の増加、カルシトリオール、副甲状腺ホルモン(PTH)、およびカルシウムによって刺激された骨組織内の**骨細胞(osteocyte)および骨芽細胞(osteoblast)**から産生されるホルモンです(図2)。FGF-23は腎臓、副甲状腺、心臓、骨、さらには他の臓器にも作用すると考えられています[71–80]。
FGF-23は、FGF-19およびFGF-21とともに、内分泌型FGFサブファミリーに属します[81]。FGFは4種類のFGFチロシンキナーゼ受容体(FGFR1~4)を介してシグナルを伝達し、RAS–MAPK経路およびPI3K–AKT経路を活性化します[82]。古典的なFGF-23シグナル伝達には、細胞外にグルクロニダーゼ活性を有する膜貫通型タンパク質であるα-Klothoが共受容体として必要であり、FGF受容体1c(FGFR1c)への結合を介して作用します[83,84]。一方で、FGF-23はKlotho非依存的にFGFR3およびFGFR4を活性化することでも作用し得ます[80,85]。
FGF-23の主な作用は、カルシトリオール産生の抑制を介して、腎臓および腸管におけるリン吸収を低下させ、血清リン濃度を正常範囲に維持することです[86]。FGF-23はリン恒常性の中心的調節因子であり、1,25-ジヒドロキシビタミンD(1,25(OH)₂D、カルシトリオール)に対する拮抗ホルモンとして機能します。
腎尿細管におけるFGF-23のリン排泄促進作用はKlotho依存性です。腎近位尿細管細胞では、FGF-23がFGFRおよび共受容体であるKlothoに結合し、管腔側膜に存在するナトリウム・リン共輸送体Na/Pi IIa(およびおそらくNa/Pi IIc)の発現を低下させます[87,88](図3)。
さらにFGF-23は、近位尿細管において25(OH)Dを活性型の1,25(OH)₂Dに変換する酵素である**1α水酸化酵素(CYP27B1)の発現を抑制し[89]、一方で25(OH)Dおよび1,25(OH)₂Dを不活性代謝物へ変換する24水酸化酵素(CYP24A1)**の発現を増加させます[90–92]。その結果、腎臓におけるカルシトリオール産生は低下し、最終的に血清リン濃度が低下します。
FGF-23の上昇は、血清カルシウム、リン、PTHに変化が現れる前から検出可能な、CKD-MBDの早期バイオマーカーです[54,55]。FGF-23濃度は末期腎不全(ESKD)発症の約5年前から上昇し始め、ESKD移行まで急速に増加し続けます[55]。FGF-23の進行性上昇は、CKD後期まで血清リン濃度を正常に保つのに寄与する一方で[54,76,93,94]、カルシトリオール産生抑制を介して続発性副甲状腺機能亢進症を促進します。
FGF-23の直接的な刺激因子としては、リポカリン2(lipocalin 2:LCN2;炎症マーカーであり鉄輸送体)[95]、低酸素状態、鉄欠乏、そしてEpoが挙げられます[96](図2)。
複数のCKD患者を対象とした臨床研究により、FGF-23の上昇は不良な予後と関連することが示されています[97–100]。さらに、Klotho欠乏は、FGF-23と死亡率あるいは心不全による入院との関連を交絡する因子ではないことが示されています[101]。
近年の科学的文献からは、カルシウム・リン代謝を制御する機構と赤血球造血を調節する機構との間に、ますます強い関連性が存在することが明らかになっています。後述するように、FGF-23は貧血および全身性炎症を促進する因子としても機能します。
2.4. ビタミンDと貧血
慢性腎臓病(CKD)患者において、ビタミンD欠乏は骨密度低下を伴う骨・ミネラル代謝異常(MBD)の進行を加速し[102–104]、さらに貧血[105–107]、心血管イベント[108]、および死亡率の上昇[108,109]と関連していることが報告されています。
in vitro研究により、マクロファージの細胞膜上に**ビタミンD受容体(vitamin D receptor:VDR)**が存在することが示されています。VDRが活性化されると、炎症性サイトカインの産生が抑制され、抗炎症性サイトカインの放出が促進されることで、赤血球系前駆細胞の増殖が促されます。したがって、CKDにおけるビタミンD欠乏は、慢性的な炎症状態を維持し、無効造血を引き起こす主要因の一つである可能性があります。
天然型および活性型のビタミンD、ならびにパラカルシトール(選択的VDR活性化薬)の使用は、炎症を軽減し、CKD患者における貧血を改善する可能性があり、その結果として同等の造血効果を得るために必要なEpo投与量を減少させ得ると考えられています[110]。
多くの臨床研究では、ビタミンD補充が赤血球造血刺激因子(erythropoiesis-stimulating agents:ESAs)への反応性を改善する可能性が示唆されています[111]。これらの効果は、ビタミンDが有する免疫調節作用およびヘプシジン低下作用に起因すると考えられます。例えば、健常人を対象とした研究では、高用量コレカルシフェロールの単回投与により血清ヘプシジン濃度が有意に低下したことが報告されています[112]。ヘプシジンは急性期タンパクであり、鉄の隔離を促進することで鉄利用能を低下させる因子です[113]。さらに、VDR関連転写因子の活性化は、赤血球系前駆細胞の増殖を促進することが示されています[114]。
一方で、無作為化比較試験では、末期腎不全(ESKD)患者においてコレカルシフェロール補充により投与3日目に血清ヘプシジン濃度が上昇したことが示されています。しかし同時に、血清1,25(OH)₂D濃度は上昇し、活性型ビタミンD製剤の必要投与量は減少しました[115]。この結果は、CKD患者におけるビタミンD補充とヘプシジン調節との関係が極めて複雑であることを示しており、最適な治療戦略を確立するためにはさらなる研究が必要であることを示唆しています。
2.5. FGF-23と鉄欠乏
慢性腎臓病(CKD)患者において、鉄欠乏は必ずしも血中の総鉄量の低下を意味するわけではなく、赤血球造血において鉄を有効に利用できない状態、すなわち機能的鉄欠乏(functional iron deficiency)を引き起こします。これは主として、CKDに特徴的な慢性炎症状態が、造血のための鉄利用を阻害するためです。
常染色体優性低リン血症性くる病(autosomal dominant hypophosphatemic rickets:ADHR)患者に関する初期の観察研究から、鉄欠乏がFGF-23合成を調節している可能性が示唆されました。ADHRはFGF-23高値を特徴とし、その結果として低リン血症および骨軟化症/くる病を呈します[116]。女性ADHR患者では、**月経や妊娠といった鉄欠乏を伴いやすい状態に一致して低リン血症の増悪(フレア)**が生じることが観察されており、FGF-23産生の増加が示唆されています[116]。
スウェーデンにおける高齢男性CKD患者を対象とした研究では、低鉄状態が腎機能や炎症とは独立して血清FGF-23高値と関連していることが示されました[117]。この関連は、CKD患者を対象とした大規模前向き研究[118]や腎移植患者(kidney transplant recipients:KTRs)[119]においても報告されています。
鉄欠乏は、HIF-1αを安定化させることで、プロタンパク質変換酵素であるフリン(furin)の発現を亢進させます。フリンはインタクトFGF-23(iFGF-23)を切断し、C末端切断フラグメント(c-FGF-23)を産生します[120,121]。このため、鉄欠乏はFGF-23の産生を増加させるだけでなく、その切断も促進します[122]。これら切断フラグメントの役割や機能については、CKD患者においていまだ十分に解明されていませんが、生理活性を有するのはiFGF-23であり、その高値はCKD患者における不良な予後と関連しています[123]。
CKDでは、いくつかの因子がFGF-23転写を亢進させ、インタクト型FGF-23の増加につながります。これらの因子の一つである鉄欠乏は是正可能であり、FGF-23産生低下につながる可能性があります。そのため、鉄補充療法はFGF-23制御の観点からも治療的意義を有すると考えられます。
血液透析患者の鉄欠乏性貧血を対象とした前向き無作為化研究では、経口鉄および静注鉄の効果が比較されました。その結果、血清cFGF-23濃度は両群で低下しましたが、血清iFGF-23濃度は静注鉄群で上昇しました。これは、経口鉄の方がiFGF-23増加を防ぐ点で有利である可能性を示唆しています[124]。
スクロフェリックオキシ水酸化鉄やクエン酸第二鉄などの鉄含有リン吸着薬による治療は、血清FGF-23濃度を低下させることが示されています。スクロフェリックオキシ水酸化鉄は血液透析患者において血清FGF-23濃度を有意に低下させ[125]、クエン酸第二鉄も血清リン濃度の低下と鉄指標の改善を伴ってFGF-23を低下させることが報告されています[126]。
同様に、ADHR患者においては経口鉄補充によりFGF-23高値が改善しました[127]。一方で、カルボキシマルトース骨格を有する一部の静注鉄製剤では、iFGF-23濃度が上昇することが示されています[128]。
マウスのCKDモデルにおいては、EpoおよびHIF-PHD阻害薬を用いて鉄利用を改善することで、iFGF-23濃度が有意に低下しました。これは、CKD患者における鉄管理がミネラル代謝の改善にも寄与し得ることを示しています[129]。
2.6. FGF-23とEpo
腎性貧血は、Epo産生低下、骨髄抑制、赤血球寿命の短縮、鉄代謝障害、慢性炎症など、複数の要因によって引き起こされます[130]。
貧血は、慢性腎臓病(CKD)患者の日常生活動作、心血管系の健康状態、さらには全体的な予後に重大な影響を及ぼします[131,132,133–135]。30年以上前に遺伝子組換えヒトエリスロポエチン(recombinant human Epo:rhEpo)が開発されたことにより、CKDにおける貧血治療は大きく変革されました[136]。しかし、rhEpoが血清FGF-23濃度に及ぼす影響は複雑かつ多面的です。
EpoがFGF-23産生を生理的に調節している可能性は、急性出血モデルマウスを用いた研究で検討されました。急激かつ重度の循環血液量減少により組織虚血が生じると、Epo産生が刺激され、骨髄における赤血球系細胞の増殖および未熟赤血球前駆細胞の末梢血への放出が起こりました。これらのマウスでは、出血後6時間以内にEpo濃度の上昇とともに、血漿中C末端FGF-23フラグメント(cFGF-23)の上昇が認められました[137]。
最近の研究においてDaryadelらは、健常マウスにrhEpoを投与すると、24時間以内にcFGF-23が急性に上昇し、数日後にはインタクトFGF-23(iFGF-23)も増加することを示しました。この急性FGF-23上昇は、予想に反して、血清リン濃度やPTH濃度の低下を引き起こしませんでした。FGF-23 mRNA発現は、骨芽細胞や骨細胞ではなく、主として赤血球系前駆細胞を含む造血骨髄で認められました。一方、同じマウスに組換えFGF-23を投与すると、腎臓におけるEPO転写産物の発現が低下し、負のフィードバック機構の存在が示唆されました[138]。
FGF-23とEpoの直接的な関係は、これ以前にも報告されています。野生型マウスにおいて、組換えヒトFGF-23の投与は血清Epo濃度を低下させ[139]、FGF-23阻害ペプチドの投与は血清Epo濃度を上昇させました[140]。これらの観察結果は、FGF-23がマウスにおいて赤血球造血を抑制する作用を有することを示唆しています。
一方、CKDマウスモデルでは、rhEpo投与により循環血中の総FGF-23が上昇しましたが、その増加はiFGF-23よりも顕著であり、FGF-23産生増加と同時に蛋白分解(切断)が促進されていることが示唆されました[141]。これらのモデルでは、骨髄がEpo刺激による新たなFGF-23産生源であることも明らかとなりました。
ヒトにおいても、血清Epo濃度およびrhEpo投与量は、CKD全ステージおよび腎移植後を通じて、総FGF-23濃度(ただしiFGF-23ではない)と独立して正の相関を示すことが報告されています。これらのデータは、マウスモデルで観察されたEpoによるFGF-23産生および代謝への影響と一致しています[141]。同様の結果は、CKDおよび慢性心不全を併存する患者においても認められ、外因性Epo投与がcFGF-23発現を著明に増加させることが示されています[123,142]。
したがって、炎症や鉄欠乏と同様に、EpoはFGF-23の転写および切断の両方を促進します。特筆すべき点として、Epoは骨以外の組織、すなわち骨髄においてFGF-23 mRNA発現を誘導します[137,138,141,143–145]。
この知見は、CKD患者で観察されるFGF-23高値が、他の因子と相まってEpo産生低下に寄与している可能性を支持するものです。
Epoがどのような機序で骨髄および骨におけるFGF-23転写を亢進させるのか、またEpoがなぜFGF-23の翻訳後切断を増加させるのかについては、現在のところ明らかではありません。FGF-23の翻訳後切断の制御は複雑であり、**GALNT3(N-アセチルガラクトサミニルトランスフェラーゼ)、FAM20C、フリン(furin)、PC5/6(Pcsk5)**など、複数の酵素が関与しています[146,147]。
2.7. FGF-23とESA低反応性
CKD患者、特に透析治療を受けている患者における貧血管理で最も重要な問題の一つが、赤血球造血刺激因子(erythropoiesis-stimulating agents:ESAs)に対する低反応性(抵抗性)です。この状態には複数の原因が関与しており、その中でも慢性炎症は重要な要因です。循環血中の炎症性サイトカインは、赤血球系前駆細胞の増殖を低下または抑制するとともに、肝臓由来ホルモンであるヘプシジンの合成を促進します[148]。ヘプシジンは、骨髄マクロファージ内への鉄の隔離を引き起こし、造血に利用可能な鉄を減少させると同時に、腸管からの鉄吸収を抑制します[149]。
ヒトを対象としたコホート研究において、FGF-23濃度と炎症マーカーとの正の関連が明確に示されています[150,151]。骨細胞様細胞株を用いたin vitro研究では、炎症性サイトカインがFGF-23 mRNA発現を亢進させることが確認されています[152]。さらにin vivoでは、炎症刺激がFGF-23の転写および切断の両方を増加させることが示されています[153]。マウスにおいて、外因性ヘプシジンを投与すると、骨におけるFGF-23発現が急性に増加し、循環血中の総FGF-23濃度が上昇する一方で、iFGF-23は正常範囲内に維持されました[153]。
日本透析アウトカム・実践パターン研究(Japanese Dialysis Outcomes and Practice Pattern Study)に参加した1044名の血液透析患者を対象とした大規模研究では、FGF-23が最も低い群および最も高い群のいずれにおいても、ESA低反応性と関連していることが示されました[154]。FGF-23低値の患者にもESA低反応性が認められたという結果は新規性があり、「FGF-23高値のみがESA抵抗性を引き起こす」という従来の考えを支持しないものでした[118,155]。FGF-23低値は、低栄養に伴うFGF-23産生低下を反映している可能性があり、この状態では、骨髄および骨細胞においてエネルギー節約機構が作動している可能性が示唆されます。**低栄養・炎症複合症候群(malnutrition–inflammation complex syndrome)**は、血液透析患者におけるESA低反応性と関連しています[156]。
ESA抵抗性に寄与するもう一つの因子が、続発性副甲状腺機能亢進症です。これは、高リン血症および低カルシウム血症によりPTHの合成・分泌が亢進する状態です。血中PTH濃度の上昇自体は、カルシウムおよびリンの恒常性を維持するための適応反応ですが、長期的には破骨細胞による持続的な骨リモデリングが骨髄線維化を引き起こし、造血能の低下につながります[157]。
したがって、続発性副甲状腺機能亢進症の是正は、CKD患者における貧血改善の治療戦略の一つとなります。維持血液透析患者を対象としたいくつかの研究では、カルシミメティクス療法の効果が検討されてきました。シナカルセトの投与により、治療を受けた患者ではヘモグロビン値が上昇し、ESA必要量が減少したことが報告されています[158–161]。
さらに、シナカルセトによる続発性副甲状腺機能亢進症の治療は、血液透析患者の貧血を改善するだけでなく[162]、FGF-23濃度の低下とも関連していることが示されています[163]。
2.8. FGF-23とHIF(低酸素誘導因子)
前述のとおり、**低酸素誘導因子(hypoxia-inducible factors:HIFs)**は、細胞内の酸素濃度低下に応答して転写を調節するタンパク質です。**プロリル水酸化酵素(prolyl hydroxylase:PHD)**は、酸素依存的にHIFを制御しています[164]。
**HIF-プロリル水酸化酵素阻害薬(HIF-PHD inhibitors:HIF-PHIs)**は、腎性貧血治療のために開発された新規作用機序を有する薬剤であり、内因性Epo産生を促進するとともに、造血のための鉄供給を改善します[2,165–167]。HIF-PHIの一つであるロキサデュスタット(roxadustat)は、FGF-23産生を増加させることが示されており、HIF-PHIが内因性Epo転写の誘導を介してFGF-23の発現および切断に影響を及ぼす可能性が示唆されています[138,153]。別の研究においても、HIF-PHIの使用がFGF-23発現を増加させることが報告されています[153]。
複数の研究により、これらに関与する分子機構が明らかにされており、炎症や鉄欠乏がHIF-1αを誘導すること[153]、およびHIF-1αが骨形成細胞においてFGF-23合成を増加させることが示されています[168]。さらに、HIF-1αは内因性Epo産生の誘導を介して間接的にFGF-23発現を増加させ、FGF-23の転写および切断を促進する可能性があります[137,141,143,144,169]。注目すべき点として、HIF-PHI治療後に誘導されるFGF-23の大部分はC末端フラグメント(cFGF-23)であることが示されています[144,153]。
一方で、この仮説とは異なる結果も報告されています。最近、ダルベポエチンαからロキサデュスタットへ切り替えた血液透析患者コホートを対象とした研究では、ロキサデュスタットへの切り替え後に血清Epo濃度の有意な上昇は認められなかったものの、エリスロフェロン濃度はダルベポエチンα治療時と同様に持続的に高値を示しました[170]。一方で、ロキサデュスタット治療群では、ヘプシジン-25が有意に低下し、総鉄結合能(TIBC)が有意に上昇しました[170]。
インタクトFGF-23(iFGF-23)濃度は正常範囲に維持される一方で、FGF-23の切断が促進されることにより、総FGF-23濃度は上昇します[120]。このcFGF-23増加の病態学的意義については現在のところ不明であり、FGF-23のC末端フラグメントが有する生物学的活性を解明するための今後の研究が必要とされています。
2.9. Klothoと貧血
FGF-23の腎臓における作用は、Klothoタンパクの存在に依存しており、Klothoは遠位尿細管をはじめ、副甲状腺などの他臓器にも発現しています。膜貫通型Klotho(transmembrane Klotho:tm-Klotho)の細胞外ドメインは、いくつかのメタロプロテアーゼによって切断され、可溶型Klotho(soluble Klotho:s-Klotho)が形成されます。s-Klothoは、FGF-23と類似したパラクリン作用を示しますが、その作用はFGF-23の合成とは独立しています。CKDの極めて早期段階からtm-Klotho発現の低下が認められ、それに伴いs-Klotho産生も低下します。
腎症マウスモデルにおいて、Sugiuraらは、Klotho mRNA発現および循環血中Klothoタンパク合成が有意に低下し、その結果として血清リン濃度が上昇することを観察しました。これらのマウスに組換えヒトEpo(rHuEpo)を投与すると、血清リン濃度が低下しました[171]。
rHuEpo治療を受けている腎移植患者117名(KTRs)と健常者22名を対象とした研究において、Leoneらは、KTRsのs-Klotho濃度が対照群より有意に高値であることを示しました。さらにKTRsでは、s-Klotho濃度がeGFRおよびFGF-23濃度と正の相関を示しました。17名のKTRsにおいて、rHuEpo治療を5週間中止したところ、治療継続群と比較してs-Klotho濃度が著明に低下しました。
同じ研究グループはまた、HK-2尿細管細胞を用いて、シクロスポリンおよびrHuEpo単独または併用投与時のKlotho mRNA発現および関連タンパク合成を評価しました。その結果、rHuEpoを先行投与し、その後シクロスポリンを投与した細胞では、単独投与の場合よりもKlotho mRNA発現が高値を示しました[172]。これらの研究から、EpoはFGF-23だけでなく、Klothoの合成も促進することが示されています。Sariらの研究では、ADPKD患者コホートにおいて、健常者と比較してFGF-23、s-Klotho、PTH濃度が有意に高値であることが示されました。一方、原因の異なるCKD患者では、一般にs-Klotho濃度は低下しています。ADPKD患者は、他のCKD患者と比べて平均ヘモグロビン値が高い傾向にありますが、これは嚢胞による腎血管床の圧迫に起因する低酸素状態によって転写因子HIFが過剰に活性化されるためと考えられます。HIF活性の上昇はEpo産生増加をもたらし、これが先行研究で観察されたKlotho合成増加の理由を説明する可能性があります[173]。
3. 骨・ミネラル代謝異常(MBD)の新規バイオマーカー
腎性骨疾患のタイプおよび重症度を同定するためのゴールドスタンダードは、現在も骨生検であり、これに代わる十分な精度を有する検査マーカーの組み合わせは、いまだ確立されていません。カルシウム、リン、ビタミンD、副甲状腺ホルモン(PTH)、および**骨特異的アルカリホスファターゼ(bone-specific alkaline phosphatase:BSAP)**は、腎性骨疾患の評価に用いられる主要なバイオマーカーです。
一方、骨粗鬆症評価に用いられる他の骨代謝マーカーは、CKD-MBDの評価および管理においては有用性が低いとされています。実際、骨吸収マーカーとして最も有用とされる血清C-テロペプチド(CTX)や、骨形成マーカーであるI型コラーゲンプロペプチド(PINP)の単量体型は、腎臓から排泄されるため、CKD患者では解釈が困難です[174–176]。
これに対して、腎臓から排泄されない骨代謝マーカーも存在し、透析非導入CKD患者における骨折リスク予測能が高い可能性があります。これには、BSAP、酒石酸抵抗性酸ホスファターゼ(tartrate-resistant acid phosphatase:TRAP5b;破骨細胞の細胞マーカー)、およびPINPの三量体型が含まれます[175,177,178]。
近年、Wnt–βカテニンシグナル伝達の障害がCKD-MBDの病態形成に関与している可能性が示唆されています[179]。スクレロスチン(sclerostin)とDickkopf-1(Dkk-1)は、Wntシグナル伝達を阻害する可溶性因子です[180]。スクレロスチンは骨細胞(osteocyte)によって産生され[181]、骨および軟骨における骨形成部位に発現しています[182]。一方、Dkk-1は骨芽細胞および骨細胞によって産生され、胚発生期には骨を含む多くの組織で発現します[183]。
スクレロスチンは前骨芽細胞および骨芽細胞に作用し、骨芽細胞の増殖および分化活性を低下させます[181]。CKDの進行に伴い、循環血中スクレロスチン濃度は糸球体濾過量の低下に比例して上昇し[184]、これは骨における産生増加によるものと考えられています[52]。CKDにおけるスクレロスチン合成の変化は早期から生じ、PTHやFGF-23の変化よりも先行して出現する可能性があり[185]、骨代謝回転の制御に関与しています。
血液透析患者では、スクレロスチン濃度はPTH値とは独立して無形成骨疾患の発症と正の相関を示し、PTHに対するスクレロスチン媒介性の骨抵抗性が形成されている可能性が示唆されています[186]。スクレロスチンは、特に透析患者において、PTHとともに高骨代謝状態を示す有望な指標と考えられています。一方で、Dkk-1は骨形態計測学的指標との相関を示していません[179,186]。
これまでに、血液透析患者ではスクレロスチン濃度が高く、その値はPTH濃度と負の相関を示すことが報告されています[187]。スクレロスチンと心血管疾患罹患率や死亡率との関連については、臨床試験の結果は一貫していません。小規模集団を対象とした研究では、透析患者においてスクレロスチン高値と死亡率との関連が報告されていますが[188–190]、一方で、大規模集団(NESOCAD研究)では、スクレロスチン濃度が最も低い患者群で生存率が低下していることが示されています[191]。
また、オステオプロテゲリン(osteoprotegerin)は、骨芽細胞を含むさまざまな細胞によって産生される可溶性受容体であり、CKD-MBD診断のための有効かつ早期の血清バイオマーカーとなる可能性があります[192,193]。その血中濃度は、保存期CKD患者および透析患者のいずれにおいても、死亡率の上昇と関連することが示されています[194]。
フェツインA(Fetuin-A)は肝細胞由来のタンパク質(ヘパトカイン)で、多彩な機能を有しています[195]。その機能には、骨外石灰化の抑制、カルシウムおよびリンを含むコロイド状カルプロテイン粒子の輸送、骨石灰化への関与などが含まれます[196]。血管石灰化におけるフェツインAの役割は完全には解明されていませんが、フェツインA低値は大動脈石灰化や主要な臨床イベントと相関することが示されています[197,198]。CKD患者では血清フェツインA濃度が有意に低下しており[199,200]、この集団においては、低フェツインA血症が全死亡および心血管死亡の増加と関連することが、複数の研究で示されています[201,202]。
近年、ミネラル代謝を制御する複雑な機構に関する知見が蓄積され、新たに多くの分子がこれらのプロセスに関与していることが明らかになってきました。しかし、これまで検討されてきた分子の中で、CKD-MBDに最も特徴的なバイオマーカーとして位置づけられるのはFGF-23です。前述の分子(およびその他の分子)については、骨および/または心血管系への関与の正確な役割は、生体液における測定法の精緻化も含めて、今後さらに検討される必要があります。
4. 考察(Discussion)
本ナラティブレビューでは、CKDに続発する貧血と、それに関連する骨・ミネラル代謝異常(CKD-MBD)との関係について、in vitro、in vivo、ならびにヒト/臨床研究から得られた主要なエビデンスを解析しました。CKD-MBDはCKDにおいて予後に最も重大な影響を及ぼす病態の一つであり、特に心血管合併症を主因とする死亡率の上昇と密接に関連しています[203]。
この文脈において中心的な役割を担う因子の一つがFGF-23です。FGF-23は、腎臓および腸管でのリン吸収を抑制することで、血清リン濃度を一定範囲内に維持するホルモンです[86]。その血中濃度は、カルシウム、リン、PTHの異常が出現する以前のCKD早期から上昇し[54,55]、さらに低酸素状態、鉄欠乏、Epoによってその産生が刺激されます[96]。FGF-23は貧血および全身性炎症を促進し、CKD患者において不良な予後と関連することが報告されています[97–100]。
CKDにおける貧血の主要因の一つである鉄欠乏は、高齢CKD患者[117,118]および腎移植患者[119]において、FGF-23高値と関連することが示されています。そのため、鉄欠乏の是正はFGF-23産生の低下と関連しており(図4)、一部の研究では、血液透析患者において経口鉄補充がcFGF-23およびiFGF-23の双方を低下させることが示されました[124–126]。これは、適切な鉄管理がミネラル代謝の改善にも寄与し得ることを示しています。
FGF-23の切断に関して重要な点として、Epoは骨以外の組織、特に骨髄においてFGF-23 mRNA発現を誘導することが示されています[137,138,141,143–145](図4)。
[図4]慢性腎臓病(CKD)に起因する貧血および続発性副甲状腺機能亢進症の管理において、FGF-23の主な作用とそれに基づく主要な治療的示唆
FGF-23とrhEpoとの関係は極めて複雑かつ多様であり、EpoがFGF-23産生の調節因子である可能性は、急性出血マウスモデルにおいて初めて検討されました。具体的には、出血後6時間でEpo濃度の上昇とともに血漿cFGF-23濃度が上昇しました[129]。その後の研究では、健常マウスにrhEpoを投与すると、急性期にcFGF-23が上昇し、数日後にはiFGF-23も増加する一方で、血清リンおよびPTH濃度は低下しないことが報告されました。これは、FGF-23産生が主として骨芽細胞や骨細胞ではなく造血系細胞で起こるためと考えられています。
さらに、同じマウスに組換えFGF-23を投与すると、腎臓でのEPO発現が低下し、負のフィードバック機構の存在が示されました[138]。CKDマウスでは、rhEpo投与により循環血中総FGF-23が上昇し、その増加はiFGF-23よりも顕著であったことから、FGF-23産生増加と同時に蛋白分解(切断)が促進されている可能性が示唆されました[141]。これらのデータは、骨髄がEpo刺激による新たなFGF-23産生源であることを示しています。
同様の結果はヒトにおいても観察されており、血清Epo濃度およびrhEpo投与量は、iFGF-23ではなく総FGF-23濃度と独立して正の相関を示しました[123,142]。したがって、炎症や鉄欠乏と同様に、EpoはFGF-23の転写および切断の両方を促進します。特に、Epoが骨髄における非骨性FGF-23 mRNA発現を誘導する点は注目に値します[137,138,141,143–145](図4)。
これらの結果は、CKD患者で認められるFGF-23高値が、他の因子と相まってEpo産生低下を引き起こし、その結果として貧血の発症に寄与している可能性を支持するものです。
貧血に影響を与えるもう一つの重要な因子が、ESA低反応性(低感受性)です。ヒトコホート研究において、FGF-23濃度と炎症マーカー(ESA反応低下の既知の要因)との関連が示されています[150,151]。興味深いことに、血液透析患者の大規模集団では、FGF-23高値のみならず低値もESA低反応性と関連していました[154]。これは、「FGF-23高値のみがESA抵抗性を引き起こす」という従来の考えに反する結果です[118,155]。
ESA抵抗性に寄与するもう一つの要因として、続発性副甲状腺機能亢進症が挙げられます。破骨細胞による骨リモデリングが持続すると、骨髄線維化を介して造血能が低下するためです[157]。続発性副甲状腺機能亢進症に対するシナカルセト治療は、ヘモグロビン値の上昇[158–162]と関連し、さらにFGF-23濃度の低下とも関連していました[163](図4)。
一方で、HIF-PHIとFGF-23の関係については結果が一貫していません。当初、ロキサデュスタットは(おそらくEPO転写誘導を介して)FGF-23産生を増加させると考えられていましたが、最近の研究では、血液透析患者においてダルベポエチンからロキサデュスタットへ切り替えても、Epo濃度の上昇は認められなかったことが報告されています[170](図4)。
5. 結論(Conclusions)
FGF-23は、CKD関連骨・ミネラル代謝異常(CKD-MBD)と貧血の双方に関連しており、これらの病態を結びつける重要な連結因子である可能性が示唆されます。FGF-23の調節機構は非常に複雑かつ多様であり、転写レベルおよび転写後レベルの両方で制御され、骨、ミネラル、腎臓に関連する因子が関与しています。さらに、鉄欠乏、Epo、炎症といったミネラル以外の因子も、FGF-23の産生および代謝に影響を及ぼします。
6. 今後の展望(Future Directions)
これらの因子がどのようにしてFGF-23転写を亢進させるのか、またFGF-23フラグメントの上昇が持つ生理的・病理的意義については、さらなる研究が必要です。これらの機序を理解することで、CKD-MBDおよび貧血の管理に新たな視点がもたらされ、患者予後の改善につながる可能性があります。


コメント